
With the plethora of molecular information coming from genetic and proteomic studies, the demand for functional analysis of the identified gene products is overwhelming. The validity of microscopy-based methods for functional studies is that single cells and even single organelles can be analyzed, which yields unique and indispensable information for understanding molecular function within the context of the cell. Protein location is an important parameter for protein function and structure is unequivocally linked to function. When microscopy-based research is combined with genetic, proteomic, molecular and biochemical approaches, a full physiological ‘picture’ of cell life will ultimately be in reach. The Cell Microscopy Center harbors a combination of state-of-the-art microscopy techniques.
Conventional electron microscopy
When the resolution of the light microscope is not enough and no knowledge about intracellular molecules is required, basic electron microscopy is the technique of choice. After in initial aldehyde fixation and post-fixation with osmiumtetraoxide the biopsies, tissues or cultured cells are embedded in an epoxy resin. This allows ultrathin sectioning of 50-70 nm thick sections. After additional staining with uranium and lead salts the ultrastructural morphology can be investigated in the electron microscope.
Immuno-electron microscopy (immuno-EM): the cryo-immunogold technique
The most widely used approach to detect proteins in cells is immuno-electron microscopy. Many protein-sorting events that determine live or death in a cell occur in 40–60-nm, often coated, membrane subdomains, which are below the detection level of the light microscope and are only distinguishable in the EM under the most favorable conditions. The Tokuyasu cryosectioning technique has been improved and perfected in the Cell Microscopy Center so that it now generally surpasses other techniques in membrane visibility and labeling sensitivity. The method includes that cells are chemically fixed, after which they are frozen and cut into 50-100 nm cryosections. The sections are thawed and labeled with antibodies that specifically recognize the molecule of interest, followed by gold particles that are detectable in the EM.
Electron tomography
Ultrathin sections (50-70 nm) provide an excellent way to observe the interior of a cell. However the thickness of a section diminishes the chance that 3D relations between organelles are found. Using thicker sections (200-300 nm) this chance is incresed. When such a section is rotated from +65 to -65 ° in an a 200KV electron microscope and a picture is taken every degree, the 3D ultrastructure of an area of interest can be visualized using special software.
Live-cell Imaging
During the past few years the use of green fluorescent protein (GFP) to study dynamic processes in living cells has led to a revival of fluorescence microscopy in life-science research. Because GFP and its variants are genetically encoded and emit visible fluorescence without the need for cofactors, they can be tagged to almost any protein to reveal in living cells its real-time kinetics and directionality of intracellular transport. Live-cell imaging provides an exceptionally powerful method to study dynamic intracellular systems.
Correlative live-cell imaging and immuno-EM
Live-cell imaging reveals the dynamics of protein traffic, yet lacks the structural resolution against which the fluorescent signal can be interpreted. Immuno-electron microscopy is unique in its capacity to localize molecules to their functional intracellular locations, but is limited in its interpretation of dynamic processes. To fill this gap between live-cell imaging and EM we have recently developed a correlative-microscopy method by which a fluorescent protein followed by live-cell imaging at any time during its itinerary can be processed for visualization by immuno-EM: by incubating cryosections with antibodies against e.g. GFP, we gold-label the previously fluorescent molecule. The advantages of this correlative technique are numerous. For example, by using antibodies conjugated to different gold sizes (e.g. 5, 10 and 15 nm), we can co-localize the formerly fluorescent protein with relevant marker-, machinery- or cargo-molecules, yielding crucial information on the structural-molecular environment of the protein.
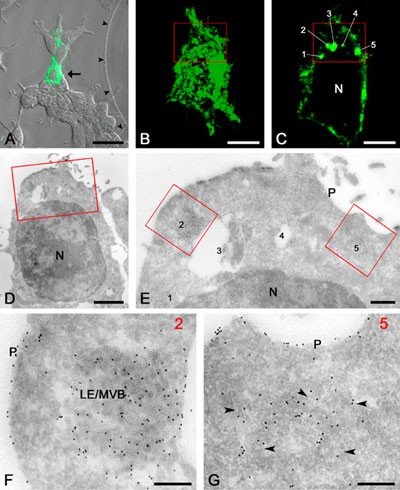
An example of correlative live-cell imaging and IEM of a single cell. A. Fluorescent image of the selected cell (arrow) that is expressing the GFP-conjugated LMP CD63. B. Fluorescent image of the entire selected cell. C. Confocal slice of the selected cell showing five distinct fluorescent spots (1-5). D. Ultrathin cryosection of the selected cell. The boxed area is the same as in B and C. E. High magnification EM of the boxed area that is shown in B-D. The numbers refer to the spots shown in C. Note the presence of 10-nm gold particles (black dots) that were obtained by immuno-labeling of GFP. F. The original fluorescent spot 2 corresponds to a LE/MVB. G. The original fluorescent spot 5 corresponds to a cluster of vesicular profiles (arrowheads) that by serial sectioning were identified as belonging to the Golgi area. N, nucleus; P, plasma membrane. The bars represent: A, 25 mm; B and C, 5 mm; D, 2 mm; E, 500 nm; F and G, 200 nm. (Adapted from Koster AJ, Klumperman J (2003), Electron microscopy: stretching the resolution limits of imaging, Nat Rev Mol Cell Biol, ss6-ss10.)
Section light – electron microscopy
In the electron microscope rare events are hard to find. In the light microscope the field of view is much larger and a rare event is seen at a glance especially if this rare event involves fluorescent markers. When an ultrathin section is processed for immuno-electron microscopy (on a special grid with location markers) it is possible to view the section on the grid in a sensitive light microscope and locate the fluorescence. After further processing the grid it can be investigated in the electron microscope. The cell of interest is located quick using the grids location markers.
High pressure freezing (HPF)
Chemical fixation may induce some ultrastructural artifacts due to the diffusion time of the fixatives into the tissues and cells. To prevent this, fresh samples are first frozen and then special ways of chemical fixation are applied. The freezing is done under high pressure so that ice crystal formation is prevented in several hundreds of microns thick cell layers. These vitreous frozen samples can then be chemically fixed in two ways:
The section fixation: 50-100 nm cryosections are cut at about -150 °C and thawed in the chemical fixative. The diffusion time for the fixative is cut back to a minimal fraction of the time needed for tissue or whole cell fixation.
Fixation after cryosubstitution: While still frozen, water in the samples is replaced by acetone, in which the fixatives are solved. At low temperature the natural structure and molecule distribution in the samples is maintained until the fixation process is finished. These cryo-substitution fixed samples can then be embedded in plastics for structural studies, or rehydrated and sectioned as in the Tokuyasu technique for immunolabeling to study molecular distributions.
Vitreous samples can also be viewed immediately in then electron microscope using a cryostage.